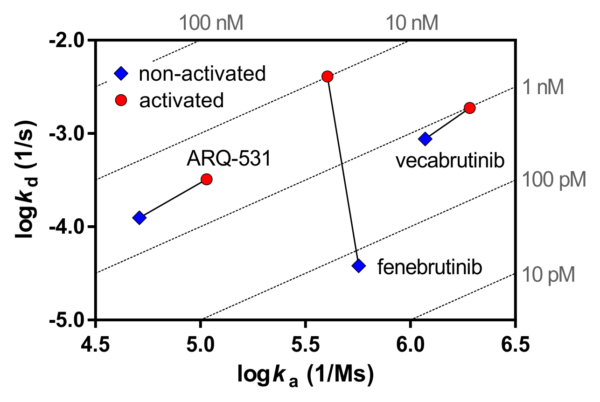
Phosphorylation state-dependent binding kinetics of BTK inhibitor fenebrutinib
Elucidating the selectivity of inhibitors for specific kinase phosphorylation states
References
Dinh et al. (2007) Activation mechanism and steady state kinetics of bruton’s tyrosine kinase, The Journal of Biological Chemistry, 282 (12):8768-8776.
Crawford et al. (2018) Discovery of GDC-0853: a potent, selective, and noncovalent bruton’s tyrosine kinase inhibitor in early clinical development, Journal of Medicinal Chemistry, 61 (6):2227-2245.
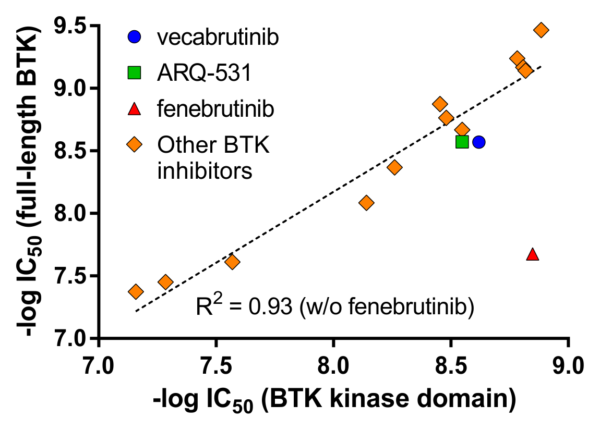
Correlation between BTK inhibitor IC50 values measured on the BTK kinase domain (residues 389–659) versus the full-length BTK enzyme.
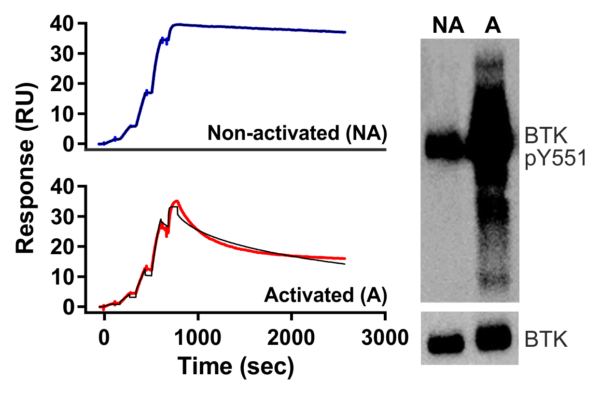
Binding curves of fenebrutinib on activated and non-activated BTK fitted with the 1:1 binding model using the Biacore T200 Evaluation software (left), and immunoblot analysis of the phosphorylation state of the BTK preparations (right).
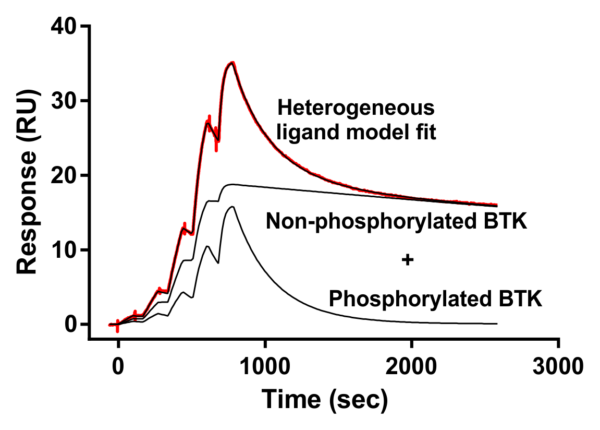
Alternative binding model for fenebrutinib on activated BTK, assuming that the enzyme preparation is heterogeneous in phosphorylation state.